
Editorial: Hospital Metropolitano
ISSN (impreso) 1390-2989 - ISSN (electrónico)2737-6303
Edición: Vol. 29 Nº 2 (2021) Abril - Junio
DOI: https://doi.org/10.47464/MetroCiencia/vol29/2/2021/64-68
URL: https://revistametrociencia.com.ec/index.php/revista/article/view/180
Pág: 64-68
Duval Alejandro Borja Menéndez1 , Carlos Xavier Erazo Santos2,
, Carlos Xavier Erazo Santos2,
Karolina Anabelle Borja Menéndez3, Mayra Consuelo Molina Herrera4,
Juan Eduardo Sánchez Núñez5
Urólogo, Universidad Central del Ecuador, Quito, Ecuador1
Urólogo, Hospital IESS San Francisco, Quito, Ecuador2
Médico, Ministerio de Salud Pública del Ecuador, Quito, Ecuador3
Médico Residente de IV año, Posgrado de Urología, Universidad Central del Ecuador, Quito, Ecuador4
Urólogo, Hospital General de México Dr. Eduardo Liceaga, Ciudad de México, México5
Introducción: El leiomiosarcoma renal es un tumor urológico raro, representa el 0,12% de los tumores renales. La detección de la masa renal está dada por estudios de imagen, pero el diagnóstico de la estirpe tumoral es histopatológico con técnicas de inmunohistoquímica. El tratamiento estándar es la nefrectomía radical; el papel de la terapia adyuvante mediante quimioterapia y radioterapia aún no está bien definido. Objetivo: Proporcionar información actualizada sobre el diagnóstico y tratamiento del leiomiosarcoma pleomórfico renal primario. Materiales y métodos: Reporte de caso y revisión bibliográfica de 11 artículos científicos encontrados en las bases de datos Medline, Scielo y PubMed. El criterio de búsqueda empleado consistió en los siguientes términos Mesh: Leiomiosarcoma renal, tumor mesenquimatoso, tumor renal. Reporte de caso: Paciente de 34 años que debuta con dolor lumbar, en estudios de imagen se evidencia masa renal izquierda, se realiza nefrectomía radical cuyo resultado histopatológico reportó leiomiosarcoma renal pleomórfico. Resultados: la urotomografía simple y contrastada es el método diagnóstico específico para la detección de masas renales; la histopatología y la inmunohistoquímica aportan información sobre la estirpe tumoral. Conclusiones: El leiomiosarcoma renal es un tumor renal raro y de mal pronóstico, su tratamiento de primera línea es la nefrectomía radical, es un tumor con alto riesgo de recidiva local y a distancia, se considera un tumor sensible a la quimioterapia por esto es considerada una terapia adyuvante.
Palabras claves: Leiomiosarcoma renal, tumor mesenquimatoso, tumor renal.
Introduction: Renal leiomyosarcoma is a rare disease, it constitutes 0,12% of malignant renal tumors, the renal mass is identified by imaging studies, but the diagnosis is given by histopathology and immunohistochemistry. The standard treatment is radical nephrectomy and the role of chemotherapy and radiotherapy is not yet well defined. Objective: To provide updated information on the diagnosis and treatment of primary renal pleomorphic leiomyosarcoma. Materials and methods: Case report and bibliographic review study of 10 scientific articles found in the Medline, Scielo and PubMed databases. The search criteria used consisted of terms: Renal leiomyosarcoma, mesenchymatous tumor, renal tumor Case report: 34-year-old patient with low back pain, which in imaging studies shows a left renal mass, a radical nephrectomy is performed. The histopathological study reported pleomorphic renal leiomyosarcoma. Results: Simple and contrasted uro-tomography is the specific diagnostic method for the detection of renal masses; histopathology and immunohistochemistry provide information on the tumor lineage. Conclusions: Renal leiomyosarcoma is a rare and aggressive renal tumor, its first-line treatment is radical nephrectomy; it is a tumor with a high risk of local and distant recurrence, it is considered a chemo-sensitive tumor, thus chemotherapy is considered an optional therapy.
Keywords: Renal leiomyosarcoma, mesenchymatous tumor, renal tumor.
| IDs Orcid | |
| Duval Alejandro Borja Menéndez: | https://orcid.org/0000-0002-2068-1697 |
| Carlos Xavier Erazo Santos: | https://orcid.org/0000-0001-7402-9664 |
| Karolina Anabelle Borja Menéndez: | https://orcid.org/0000-0002-7028-8033 |
| Mayra Consuelo Molina Herrera: | https://orcid.org/0000-0003-2671-9833 |
| Juan Eduardo Sánchez Núñez: | https://orcid.org/0000-0002-2013-8248 |
| Correspondencia: | Md. Duval Alejandro Borja Menéndez |
| Teléfonos: | 984044163 |
| e-mail: | duvalborja@hotmail.com |
INTRODUCCIÓN
Los tumores renales constituyen el 3% de los tumores sólidos del adulto y el carcinoma de células claras es el subtipo más frecuente (75 al 80%)1; los sarcomas son tumores que derivan del mesodermo embrionario2,3. En Estados Unidos, en el año 2005, se diagnosticaron aproximadamente 10.000 nuevos casos de sarcomas, de los cuales solo el 2,1% correspondió al tracto genitourinario4. El leiomiosarcoma es el subtipo histológico más común, siendo el 60 al 70% de todos los sarcomas de riñón. Se presenta entre los 50 y los 60 años de edad5. La nefrectomía radical es el estándar de oro para el tratamiento de masas renales, que puede ser complementado con quimioterapia y radioterapia6. Para su diagnóstico se requieren técnicas de histopatología e inmunohistoquímica que confirmen su histogénesis7. El leiomiosarcoma con un gran componente pleomórfico se denomina leiomiosarcoma pleomórfico8. Los sarcomas renales son tumores muy agresivos por la tendencia a la recurrencia local y a distancia, además de producir metástasis por vía hematógena6,9. El objetivo de la presente revisión es proporcionar información actualizada sobre el diagnóstico y manejo clínico-quirúrgico del leiomiosarcoma pleomórfico renal primario.
METODOLOGÍA
Estudio de reporte de caso y revisión bibliográfica con análisis sistemático de 10 artículos científicos encontrados en las bases de datos Medline, Scielo y PubMed, cuyas fechas de publicación corresponden a los últimos 5 años. El criterio de búsqueda empleado consistió en los siguientes términos Mesh: Leiomiosarcoma renal, tumor mesenquimatoso, tumor renal.
REPORTE DE CASO
Paciente masculino de 34 años, soltero, de ocupación comerciante, grupo sanguíneo ORh+, sin antecedentes clínicos de importancia, que debuta con dolor lumbar izquierdo; se realiza ecografía y tomografía abdominal, identificándose una masa renal izquierda (Figura 1 y 2).
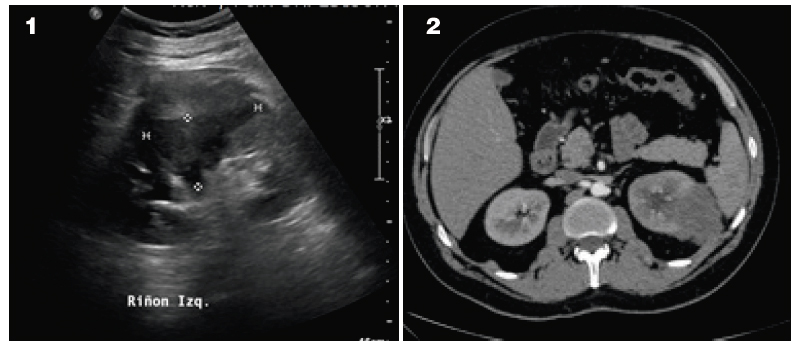
Figuras. 1) Ecografía renal: lesión hiperecogénica de 57 x 78 x 67 mm, de bordes irregulares, localizada en el segmento medio del parénquima renal izquierdo. 2) Tomografía que revela una formación intra y extraparenquimatosa en el riñón izquierdo, de bordes lobulados e imprecisos, que se extiende fuera de los límites capsulares y contacta con planos tisulares de la pared abdominal contigua y el bazo.
Se realiza nefrectomía radical izquierda por lumbotomía; se observa la presencia de una masa renal vascularizada de aproximadamente 8 x 8 cm de diámetro, de consistencia dura, en el segmento lateral del riñón, en estrecha cercanía con el bazo. El reporte histopatológico revela un leiomiosarcoma de alto grado, con áreas pleomórficas y necrosis menor del 50% del tumor, bordes quirúrgicos libres de células tumorales (Figura 3).
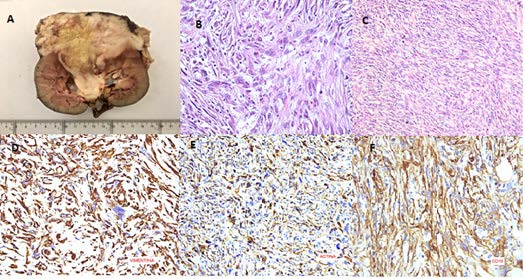
Figura 3. A) Masa renal izquierda de 9,5 x 7,6 x 7 cm que compromete la cortical, pelvis renal, hilio y tejido adiposo perirrenal en un espesor de 3 cm. B) Hematoxilina Eosina 40X, marcado pleomorfismo nuclear, multinucleación, nucléolos grandes, figuras de mitosis. C) Hematoxilina Eosina 10X, neoplasia fusocelular, disposición en haces paralelos, abundante celularidad, pleomorfismo, hipercromatismo, alto índice núcleo-citoplasmático, mitosis dispersas. D) Vimentina 40X, positiva citoplasmática. E) Actina 40X de músculo liso positivo para células neoplásicas. F) CD 10 40X, positivo en membranas de células neoplásicas.
En una tomografía de seguimiento después de 3 meses de la nefrectomía, se evidencia nódulos pulmonares bilaterales sugestivos de metástasis a distancia. En el lecho quirúrgico se evidencia recidiva local con presencia de masa con densidad de tejido blando; no se observa un adecuado plano de clivaje con la pared abdominal posterior ni con la región inferior del bazo. Adyacente al músculo psoas izquierdo, se evidencian dos imágenes de aspecto lobulado de 24 x10 x13 mm que sugieren ser implantes orgánicos, y, en la pared lateral izquierda del abdomen, adyacente al músculo transverso, se observa una imagen nodular de 15 x13 mm sugestiva de secundarismo (Figura 4). Se inicia tratamiento paliativo de quimioterapia con protocolo de ifosfamida y doxorrubicina por 6 ciclos. El paciente sufre una fractura patológica del húmero proximal, y finalmente fallece un año posterior al diagnóstico por neumonía adquirida en la comunidad y paro cardiorrespiratorio.
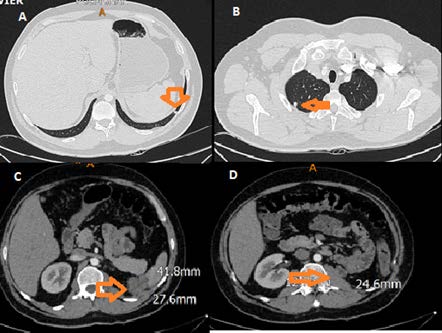
Figura 4. A) Micronódulo de 5 mm en base pulmonar izquierda. B) Micronódulo localizado en el pulmón derecho. C) Tomografía computarizada contrastada de abdomen: recidiva local. D) Tomografía computarizada: recidiva local.
DISCUSIÓN
Los sarcomas son tumores que derivan del mesodermo embrionario y su localización más común es estómago, intestino y retroperitoneo. El leiomiosarcoma renal es uno de los subtipos de tumores renales más raros, constituye apenas el 0,12% de los tumores renales malignos7 y fue descrito por primera vez en el año 1919 por Berry9,10. El leiomiosarcoma es un tumor más frecuente en mujeres, con una relación 2:1. La causa exacta no es completamente conocida, pero varios estudios sugieren una asociación con genes localizados en el cromosoma X. El leiomiosarcoma puede encontrarse por igual en ambos riñones o de manera unilateral9.
Los pacientes frecuentemente debutan con dolor lumbar, hematuria y masa abdominal, por lo que es difícil diferenciar entre un leiomiosarcoma renal y un tumor de células claras mediante técnicas de imagen; por lo tanto, su diagnóstico suele ser postoperatorio10. Debido a que el sarcoma renal es una patología rara, no se ha establecido un tratamiento estandarizado. La cirugía es la mejor opción curativa, por lo que la nefrectomía radical es el estándar de oro en el tratamiento de masas renales de características tumorales9,10. La guía europea y la guía de la NCCN (National Comprehensive Cancer Network) sobre el carcinoma de células renales, en su actualización del año 2021, indica que el tratamiento quirúrgico es la primera línea en el manejo de pacientes con masas renales, sin distinción de la estirpe tumoral, como terapia curativa. En pacientes con tumores renales localmente avanzados, el tratamiento quirúrgico se mantiene como la primera opción terapéutica, aunque puede ser complementado con quimioterapia adyuvante11,12.
La histogénesis del leiomiosarcoma es desconocida, pero la hipótesis más aceptada es la que considera las células de la cápsula renal, vasos sanguíneos y músculo de la pelvis renal como sus precursores6,9. Histológicamente, el leiomiosarcoma presenta gran cantidad de mitosis, necrosis focal y pleomorfismo celular6; la atipia nuclear es prominente. En la inmunohistoquímica, el tejido tumoral es positivo para vimentina y marcadores de músculo liso6. Para el diagnóstico de leiomiosarcoma pleomórfico, el punto de corte del componente pleomórfico debe tener una relación 2:3 en relación al volumen tumoral total, siendo el leiomiosarcoma pleomórfico más agresivo. La correlación entre la extensión del componente pleomórfico y el pronóstico todavía no es clara3,8. El patrón de crecimiento de los leiomiosarcomas es fascicular, con formación de bandeletas que dan la apariencia macroscópica de ángulos6.
Un estudio sobre el leiomiosarcoma con 33 pacientes, demuestra que la media de sobrevida fue de 3,9 años, el 73% de pacientes sobrevivieron a los 2 años, pero solo el 38% sobrevivieron a los 5 años; 20 de los pacientes murieron a causa de la enfermedad. Estos datos demuestran el mal pronóstico de esta enfermedad8.
Para hacer un diagnóstico de sarcoma renal primario se deben cumplir los siguientes criterios10: a) el paciente no debe tener un sarcoma en otro lugar; b) la masa debe ser compatible con un origen renal en lugar de una afectación por continuidad en el retroperitoneo; c) la variante sarcomatoide del carcinoma de células renales debe ser excluida; y, d) si existieran metástasis, éstas deben ser más pequeñas que la masa renal. El tamaño menor de 5 cm, bajo grado histológico, y enfermedad limitada al riñón, son factores asociados con resultados favorables. El grado histológico está asignado en base al conteo mitótico, necrosis y al pleomorfismo nuclear. El factor pronóstico más importante es el margen de resección libre de tumor, mientras que la diseminación a distancia en el momento del diagnóstico, es un signo de mal pronóstico9.
El papel de la quimioterapia no está aún establecido, pero este tumor se considera sensible, particularmente al esquema de doxorrubicina e ifosfamida. La terapia neoadyuvante con radioterapia estaría indicada si se reportan bordes tumorales positivos en el estudio histopatológico y cuando se trata de tumores de grado histológico intermedio y alto1,10. Cuando se presentan metástasis a distancia, la media de supervivencia es de 8 a 12 meses; el tratamiento se basa en combinar la resección de las metástasis y de la recurrencia local siempre que sea posible, además del tratamiento con quimioterapia a base de ifosfamida en altas dosis como monoterapia o en combinación1.
CONCLUSIÓN
El leiomiosarcoma renal es un tumor en extremo raro y agresivo, con alto riesgo de recidiva local y a distancia por metástasis hematógenas. El diagnóstico es principalmente postquirúrgico, ya que los estudios de imagen no cuentan con un patrón distintivo que lo diferencie de otros tumores renales. El tratamiento de primera línea es la nefrectomía radical con márgenes amplios, con bordes libres de tumor. El esquema de tratamiento adyuvante aún no está definido debido a lo raro del tumor, por lo que se han descrito varios esquemas de quimioterapia y radioterapia. El leiomiosarcoma renal es considerado un tumor sensible a la quimioterapia, por lo que se recomienda esquemas que contengan doxorrubicina e ifosfamida. El escaso número de casos impide la estandarización de protocolos de tratamiento y seguimiento definitivos para tumores renales poco frecuentes.
Financiamiento
Se trabajó con recursos propios de los autores.
Conflicto de interés
Los autores reportaron no tener ningún conflicto de interés, personal, financiero, intelectual, económico y de interés corporativo.
Agradecimientos
Dedicado a los colegas y amigos que ayudaron con esta revisión.
Cesión de derechos de reproducción
Los autores cedemos todos los derechos del presente manuscrito en caso de ser publicado por la revista Metro Ciencia.
Declaración de artículo inédito
Este manuscrito original no ha sido publicado y no está bajo consideración para publicación en otra revista.
Aporte de autores
Concepción y diseño del trabajo: DB.
Recolección y obtención de resultados: KB.
Análisis e interpretación de datos, redacción del manuscrito: DB, KB, MM, JS.
Revisión crítica del manuscrito: CE
Aprobación de su versión final: todos los autores.
REFERENCIAS BIBLIOGRÁFICAS
Borja DA, Erazo CX, Borja KA, Molina MC, Sánchez JE. Leiomiosarcoma pleomórfico renal primario: Reporte de caso y revisión de la literatura. Metro Ciencia [Internet]. 29 de abril de 2021; 29(2): 64-68. https://doi.org/10.47464/MetroCiencia/vol29/2/2021/64-68