
Doctora en Medicina y Cirugía; Especialista en Pediatría; Hospital Clínica San Agustín; Loja, Ecuador1
El síndrome de insensibilidad androgénica (SIA) es una de las anormalidades de la diferenciación sexual (desarrollo sexual diferente). Es un trastorno genético dependiente del cromosoma X, produce una alteración en el receptor de andrógenos, se asocia con testículos en las mujeres cuyo cariotipo es XY y con agenesia vaginal y uterina. Acuden a la consulta médica los padres con su hija recién nacida de 12 días de edad. Motivo de consulta: masa en la región inguinal derecha. Examen físico: signos vitales normales, activa al manejo, reactiva. Se observa una masa en la región inguinal derecha de aproximadamente 2 cm de diámetro, reductible, no dolorosa. Genitales externos femeninos: normales. La paciente es referida al Servicio de Cirugía para proceder a la corrección del defecto herniario. Se indica realizar un estudio citogenético y medir los niveles hormonales en sangre. Resultado del estudio anatomopatológico posquirúrgico, luego de 7 días de haber sido intervenida quirúrgicamente: “Tejido gonadal de tipo testicular con zonas de congestión vascular y hemorragia focal”. Los niveles hormonales sanguíneos son normales; el cariotipo es normal masculino XY. Diagnóstico: debido a que el resultado del cariotipo es concluyente, se diagnostica síndrome de insensibilidad androgénica (SIA)” completo.
Palabras claves: telangiectasia capilar, malformaciones vasculares, resonancia magnética.
Androgen insensitivity síndrome (AIS) is one of the causes of abnormalities in sexual differentiation (different sexual development). SIA is an X-linked genetic condition caused by an androgen receptor disorder, associated with vaginal and uterine agenesis, and the presence of testicles in women with an XY karyotype. Parents with 12-day-old neonates go to medical consultation. The reason for consultation is a mass in the right inguinal region. On physical examination: normal vital signs, active on management, reactive. A mass is observed at the level of the right inguinal region of approximately 2 cm in diameter, reducible and not painful. Female external genital with normal characteristics. The patient is referred for surgery to correct hernia defect. A cytogenetic study and blood hormone leves are indicated. Seven days after the intervention, parents came with the results of the postoperative pathological study: testicular gonadal tissue with áreas of vascular congestion and focal hemorrhage. Blood hormonal lever are normal and anormal XY male karyotype is seen. Diagnosis: the result of the karyotype is conclusive and a complete AIS is diagnosed.
Keywords: androgen insensitivity syndrome, inguinal hernia, karyotype.
| IDs Orcid | |
| Patricia Moreira Aguirre: | https://orcid.org/0000-0002-4522-0782 |
INTRODUCCIÓN
El síndrome de insensibilidad androgénica (SIA) se manifiesta cuando un individuo genéticamente masculino (46 XY) tiene resistencia a los andrógenos (RA). Su presentación clínica es variada, pero frecuentemente su fenotipo es femenino1. Los reportes iniciales del SIA datan del siglo XIX e incluso se supone que personajes conocidos como la reina Isabel I de Inglaterra y Juana de Arco tuvieron esta afectación. Morris, en 1953, lo describió y denominó “feminización testicular” después de analizar 82 casos descritos en la literatura; los pacientes estudiados presentaban fenotipo femenino, escaso vello corporal, amenorrea y testículos2.
El SIA es causado por la mutación del gen del receptor de andrógenos (RA). No existe déficit enzimático, carencia de andrógenos ni de hormona antimülleriana. El SIA se debe a diversas anomalías genéticas en la RA que se localizan en el cromosoma X (Xq 11-q12); más del 30% de casos son mutaciones de novo. Las expresiones fenotípicas del SIA dependen de la expresión del defecto. Se hereda por un patrón recesivo ligado al cromosoma X. El SIA se lo clasifica entre las anomalías de la diferenciación sexual (ADS)2,3.
Las ADS incluyen un grupo amplio de discordancias entre los criterios fenotípico (genital), cromosómico y gonadal que definen la diferenciación sexual. Durante el Consenso de Chicago de 2006 se las denominó “anomalías o trastornos del desarrollo sexual” o “estados intersexuales”; actualmente y debido al rechazo de esta terminología médica se propuso que se las denomine “desarrollo sexual diferente” (DSD).
Durante la vida fetal, la diferenciación sexual sigue un proceso secuencial de desarrollo regulado por varias hormonas, genes y proteínas. Durante las primeras 6 semanas (primera etapa) no hay diferencia entre ambos sexos y el embrión es pluripotencial. Desde la semana 7 se inicia la diferenciación gonadal mediante el control de múltiples genes; el gen SRY del cromosoma Y es determinante para el desarrollo testicular. La diferenciación sexual en el varón está regulada por el efecto de las hormonas testiculares; v.gr.: testosterona y antimülleriana; en cambio, en la mujer la diferenciación genital ocurre por ausencia de estas hormonas. Si ocurre alguna alteración, genética o medioambiental, en algún momento del desarrollo gonadal se presentarán anomalías (disgenesia gonadal, alteraciones de los genitales internos o externos).
El desarrollo gonadal inadecuado se manifiesta durante el nacimiento como ambigüedad sexual o discordancia entre el fenotipo y el genotipo. La pubertad puede estar atrasada, manifestar virilización insuficiente o excesiva y amenorrea. En las mujeres adultas en edad reproductiva puede presentarse infertilidad y menopausia precoz3,4.
Epidemiología del SIA
La Unión Europea cataloga a la SIA como enfermedad rara. La incidencia poblacional es de 1 a 5 casos de SIA completo por cada 100.000 recién nacidos vivos varones. La prevalencia de SIA completo no supera los 5 casos por 10.000 habitantes. Se desconoce la prevalencia del SIA incompleto o leve1,2. La prevalencia se eleva cuando se estudia mujeres que tienen hernias inguinales (de 0,8 a 2,4%)1,2,5. En pacientes con amenorrea, el SIA es el tercer diagnóstico etiológico6.
Genética del SIA
El gen del receptor androgénico (RA) pertenece a un subgrupo de factores de transcripción nuclear. La unión del andrógeno a su receptor favorece el cambio de la estructura del RA desde un estado inactivo a un activo y favorece la unión al ADN. El gen del RA se ubica en el brazo largo del cromosoma X (Xq11-q12), está conformado por 8 exones y codifica a una proteína de 114 kD (Figura 1). La porción amino-terminal de la proteína es codificada por el exón 1, que es el más largo. Esta región inicia la transcripción génica. Los exones 2 y 3 codifican el dominio de unión al ADN. Las mutaciones de la región de unión al ADN provocan que el receptor sea afuncional por incapacidad de unión al ADN, por lo cual no se inicia la transcripción génica. El exón 4 codifica la porción denominada “bisagra” que se relaciona con la configuración tridimensional de la estructura de la proteína que codifica el gen.
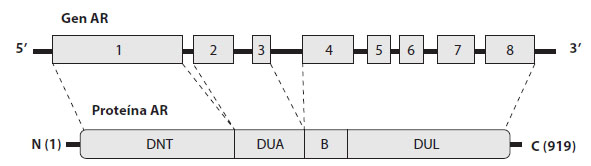
Figura 1. Estructura del gen receptor de andrógenos. El gen AR consta de 8 exones (diagrama superior) que codifican el receptor de andrógenos con un producto génico cuyo tamaño típico es de 919 aminoácidos. La proteína resultante (diagrama inferior) está compuesta por un dominio N-terminal (DNT), un dominio central de unión al ADN (DUA), una región de bisagra corta (B) y un dominio de unión a ligandos (DUL) C-terminal.
Los exones 5 a 8 codifican los ligandos, que son las porciones que requiere el receptor para unir los andrógenos al receptor. En el dominio de unión a los andrógenos es donde se ha detectado la mayoría de mutaciones.
Estas mutaciones alteran la afinidad de unión de los andrógenos y/o su especificidad de unión. Se conocen más de 800 mutaciones que se manifiestan mediante distintas enfermedades2. En aproximadamente el 40% de pacientes con SIA no hay antecedentes familiares de la enfermedad6.
Manifestaciones del SIA
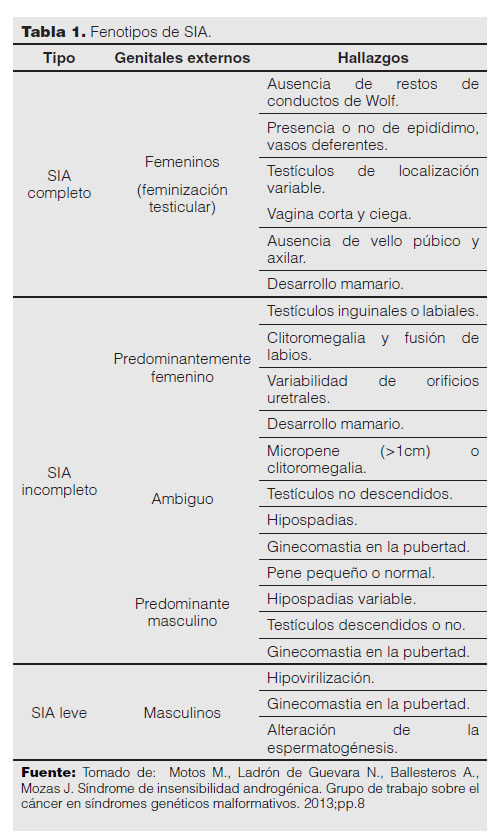
Hay 3 fenotipos de SIA:
SIA completo (síndrome de Morris).
SIA parcial (síndrome de Reinfenstein).
SIA leve o mínimo (síndrome del varón infértil)1,2,7 (Tabla 1).
SIA completo
Los pacientes con SIA completo no presentan respuesta a los andrógenos, su desarrollo genital externo femenino es completamente normal, se educan como mujeres. La inclinación sexual no se ve afectada, tampoco su identidad. La presentación clínica puede ser al nacimiento o durante la infancia por la presencia de masas inguinales o hernias en niñas completamente normales. Las gónadas son testículos azoospérmicos, su localización es variable, la mayoría de veces se encuentran en los conductos inguinales. Es muy raro que se lo diagnostique durante la infancia, a veces se realiza luego de la corrección quirúrgica del defecto herniario frente al hallazgo, por el patólogo, de tejido testicular. El diagnóstico también se puede realizar con un cariotipo prenatal6.
El patrón de crecimiento estatural en el SIA es igual al de las niñas normales; no obstante, la talla final es más cercana a la de los niños normales. El comportamiento suele ser el de una mujer normal, su desarrollo mamario es normal, el vello axilar y pubiano está disminuido o ausente6.
Es habitual que el diagnóstico de SIA se realice en la pubertad debido a la amenorrea primaria; más tarde puede haber esterilidad. En raros casos, el SIA completo puede manifestarse con clitoromegalia (masculinización de los genitales externos) o fusión de labios y desarrollo de epidídimo, conductos deferentes y vesículas seminales1,2,4,8.
SIA incompleto
Aunque es infrecuente, su presentación clínica es muy variable; incluye ambigüedad sexual y otras manifestaciones que pueden variar desde la masculinización casi normal hasta la feminización casi completa. La variabilidad de la presentación sintomática depende del tipo de mutaciones que afectan al gen RA. Al nacer o en la infancia ya se puede precisar la ambigüedad sexual. Cuando el individuo se define como mujer se presenta clitoromegalia, fusión de labios menores y vello púbico durante la pubertad1,6. Cuando se identifica como varón puede presentar micropene, hipospadias y criptorquidia; las células germinales en sus testículos son muy escasas o carecen de ellas. En la pubertad puede haber ginecomastia y aspecto eunucoide.
SIA mínimo o leve
En este grupo se pueden incluir a los varones con aspecto físico normal o signos de hipovirilización, puede haber ginecomastia ligera y también espermatogénesis leve. Durante la infancia y la pubertad se desarrollan normalmente. La fertilidad está determinada por el grado de maduración de las células germinales; algunos pueden tener hijos1,6.
Otras manifestaciones asociadas con el SIA
Puede existir otras manifestaciones más raras (endocrinas o neurológicas) que no son parte del SIA y son provocadas por alteraciones del gen AR. La enfermedad de Kennedy o síndrome de atrofia muscular espinobulbar es un desorden ligado al cromosoma X; se debe a una variación de la estructura del gen AR y se caracteriza por degeneración progresiva de las neuronas motoras anteriores asociada con ginecomastia, espermatogénesis defectuosa y perfil hormonal de resistencia androgénica1,6.
MATERIALES Y MÉTODOS
Historia clínica
Se realizó una historia clínica completa, incluyendo antecedentes familiares, antecedentes prenatales e historia patológica de padres y familia. El examen físico fue minucioso. Debido a los hallazgos del examen físico se solicitó una ecografía de la región inguinal. Luego de una semana y después de la corrección quirúrgica de la hernia inguinal, acuden con los resultados anatomo-patológicos. Se solicita exámenes hormonales LH, TSH, DHEA y testosterona, electrolitos séricos, pruebas de tamizaje neonatal y cariotipo.
Cariotipo
Se tomó una muestra de sangre periférica. Se realizó un cultivo celular compuesto por: medio RPMI, L-glutamina, antibiótico antimicótico, suero newborn y fitohemaglutinina. Se sembraron 500 uL de la muestra y se realizó el conteo de 20 metafases. Se analizó la estructura de los cromosomas por medio de la técnica de bandas G.
Diagnóstico
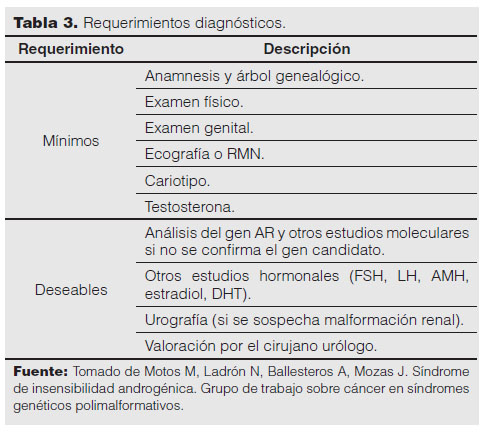
Frente a la presencia de un recién nacido con anomalía genital, la valoración del cariotipo debe ser el primer paso diagnóstico; esta es la base de la nueva clasificación en los trastornos del desarrollo sexual, acordada en el Consenso de Chicago en 2006 (Tabla 2)9,10,11. Se realizó una lista de verificación con el propósito de sustentar la sospecha de una mutación del receptor de andrógenos, que consistió en los siguientes determinantes:
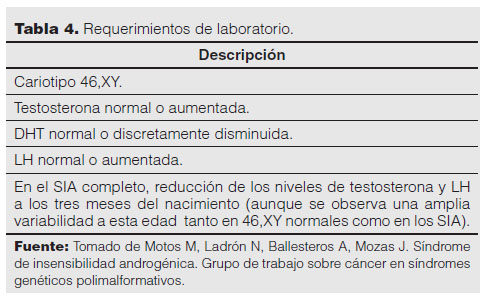
En las Tablas 3 y 4 se enumeran los criterios diagnósticos. El diagnóstico diferencial incluye: déficit de la enzima 5α-reductasa, síndrome de Mayer-Rokitansky-Küster-Hauser, síndrome de Kallmann y agenesia de las células de Leydig por anomalía del receptor de LH1.
Test bioquímicos
Se realizaron los niveles séricos hormonales de testosterona total (Chemiluminescent Microparticle Immunoassay. ARCHITECT, Abbott Laboratories), LH (inmunoensayo quimioluminiscente con el Kit de biotrol diagnostic para el autoanalizador MAGIA L) y FSH (inmunoensayo quimioluminiscente con el Kit de bioMérieux S.A. en el autoanalizador MAGIA L).
Evolución
Los problemas en estos pacientes son psicológicos, riesgo de malignización gonadal, infertilidad y osteoporosis en los adultos14. Actualmente, se está estudiando la asociación entre SIA con obesidad y la enfermedad cardiovascular. Se ha descrito que las mujeres con SIA tienen mayor resistencia a la insulina, aumento de la masa grasa y niveles de colesterol total y LDL mayores a los de la población normal15. Estos elementos, básicos para el seguimiento de la evolución del caso descrito, se transmitieron a los familiares de la paciente.
RESULTADOS
Examen físico
Neonata, peso 3,4 kg, talla 48 cm, perímetro craneal 34,5 cm, temperatura 36,8 °C, frecuencia cardíaca 125 x´, frecuencia respiratoria 38 x´, saturación 93%, fenotipo femenino, genitales externos normales, masa inguinal derecha compatible con hernia inguinal, reductible a las maniobras pertinentes.
Anamnesis
Historia personal: no hay antecedentes de consanguinidad entre los padres; tampoco patológicos familiares. Nació por parto distócico, a término, Apgar 9 y 10; alimentación: lactancia materna exclusiva. Grupo sanguíneo ORh+.
Ecografía inguinal
En el lado derecho: defecto de la pared de 8 mm de diámetro en el orificio inguinal interno, ubicados por fuera de los vasos femorales; a través orificio inguinal protruye un saco herniario hiperecogénico probablemente adiposo que mide 22 x 10 mm. Se evidencia reducción espontánea del contenido herniario durante el transcurso del estudio. Impresión diagnóstica: hernia inguinal indirecta derecha autorreductible. La paciente fue transferida a cirugía pediátrica para la corrección del defecto herniario. Resultado anatomopatológico de la pieza quirúrgica: tejido gonadal de tipo testicular con zonas de congestión vascular y hemorragia focal.
Test bioquímicos
Pruebas hormonales: TSH 5 uU/ml, DHEAS 7,1 ng / ml (0,5-19,4 ng/ ml), LH 0,5 (< 0,6 mUl / ml), androstenodiona 0,25 (0,05-0,4 ng / ml), testosterona 0,12 (0,06-0,82 ng/ ml).
Electrolitos séricos: sodio 138 mEq / L (135-145 mEq / L), potasio 3,5 mEq / L, cloro 101 mEq / L (100-106 mEq / L), calcio iónico 1 mmol / L (1-1,3 mmol/L).
Prueba de tamizaje metabólico neonatal: negativa.
Cariotipo: mediante el cultivo de linfocitos provenientes de la muestra sanguínea se pudo precisar su origen masculino (Figura 2).
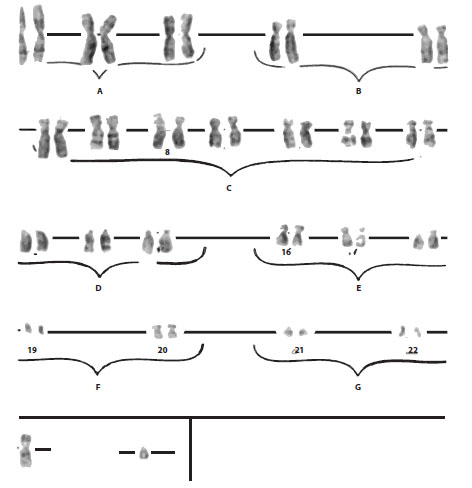
Figura 2. Cariotipo. Muestra de origen masculino: 46 cromosomas en las 20 metafases contadas.
Fuente: Los autores
DISCUSIÓN
El SIA es una patología rara y a menudo poco imaginada por nosotros, los pediatras, cuando observamos a una niña con caracteres sexuales completamente femeninos. El hallazgo anatomopatológico en la hernia inguinal nos conduce a realizar todo el proceso diagnóstico. Este caso clínico concuerda con lo referido por Hiort O10 considerando el diagnóstico clínico de SIA en mujeres con hernias inguinales o masas en los labios mayores. Según este autor, aproximadamente 1 a 2% de pacientes de fenotipo femenino con estas hernias pueden ser diagnosticadas de SIA. El SIA completo tiene una incidencia poblacional entre 1 y 5 en 100.000 personas. Al igual que Beltrán O5, el diagnóstico durante la niñez suele efectuarse por la presencia de hernia inguinal; el perfil bioquímico no refleja anormalidades; el cariotipo permite aproximarse al diagnóstico.
Los neonatos y lactantes con SIA no muestran el incremento usual de gonadotropinas y testosterona, mientras aquellos con SIA parcial sí. La hormona antimülleriana puede encontrarse en rango normal o algo elevada, lo que sugiere función testicular normal de las células de Sertoli10.
En los niños y en los adultos con sospecha de SIA completo o incompleto se debe corroborar la capacidad de sintetizar testosterona; se logra administrando hormona coriónica gonadotrópica (hCG). Hay diferentes protocolos de estimulación de hCG: 1.000 a 2.000 UI/ día/ 3 a 5 días o alternativamente 5.000 Um2 en una dosis simple; luego de 72 horas se mide los niveles séricos de androstenodiona, testosterona y dehidrotestosterona10.
Tanto en los casos referidos por Beltrán O5, Rodríguez Estévez A9, Hiort O10 y Guerrero Fernández J3, sólo la determinación molecular del defecto define el sitio exacto de la mutación.
No existe un tratamiento que prevenga o revierta el SIA. El manejo está en inicio enfocado en aconsejar a la familia y en la asignación del sexo del infante con genitales ambiguos. La asesoría para los padres debe estar guiada por un equipo multidisciplinario que incluye a endocrinólogos pediatras, cirujanos pediatras, urólogos pediatras, asistentes sociales, etc.3,10. La asignación del sexo en el SIA completo3,5,8,10 no plantea problemas porque el fenotipo es femenino y su educación debe ser concordante. El abordaje psicológico y psiquiátrico durante la niñez y adolescencia permite guiar adecuadamente a las pacientes con SIA y a sus padres o cuidadores. Luego del consentimiento de los padres y con una asesoría psicológica adecuada, se debe informar a la paciente el diagnóstico y la fisiopatología. Se debe dar lineamientos respecto de la calidad de vida; hacerlo de forma adecuada según el período etario (niñez, adolescencia y adultez). Asesorar e informar repetidamente de su capacidad de vivir como mujer, con el debido conocimiento de su infertilidad2.
Se recomienda la asesoría genética en todos los casos6,9,10 porque es una enfermedad genética recesiva ligada al cromosoma X. La paciente cuyo cariotipo es 46,XX y presenta mutación en uno de sus cromosomas es portadora asintomática (no afectada); en el caso del individuo cromosómicamente varón 46,XY, debido a que tiene solo un cromosoma X, no es portador y puede ser siempre afecto o no afecto. La madre de una persona con SIA puede ser portadora heterocigota de la mutación, su fenotipo puede evidenciar rasgos mayores que demuestren la mutación, aunque puede haber antecedentes de menarquia tardía o distribución asimétrica del vello pubiano. Riesgo de heredar el defecto: 50% hijas no portadoras y 50% de portadoras; en los hijos, 50% afectados y 50% no afectados6.
Los pacientes con SIA tienen mayor tendencia a la malignización gonadal; el riesgo es bajo antes de los 25 años, pero su frecuencia aumenta entre los 30 y 50 años. El porcentaje de malignización gonadal ocurre en 1,5 a 2% de los testículos no descendidos, y es más frecuente cuando los testículos son abdominales en comparación con los inguinales10,19. La extirpación de los testículos se recomienda en los mayores de 16 años; es decir, pasada la pubertad, tiempo en el cual la feminización es completa. Durante este lapso, son recomendados los controles clínicos y mediante imágenes; v.gr.: ecografía o resonancia magnética de las gónadas20. La extirpación en edades tempranas está indicada cuando los testículos son palpados en la región inguinal o por el aspecto estético1,10.
Otro pilar del tratamiento es la terapia de reemplazo hormonal (TRH) con estrógenos en las pacientes con SIA completo o parcial en las que se ha decidido por el sexo femenino. La TRH se inicia precozmente cuando la gonadectomía se realizó antes o durante la pubertad. La edad y dosis inicial del tratamiento hormonal depende del percentil de crecimiento, velocidad de crecimiento, edad ósea, talla y predicción de talla adulta10. En las mujeres adultas, la TRH debe empezar a admininistrarse inmediatamente después del diagnóstico10,17. Cuando existe acortamiento vaginal se realiza cirugía o técnicas de dilatación que evitan la dispareunia y permiten las relaciones sexuales16,17. En los pacientes que tienen genitales ambiguos, el tipo y número de cirugías depende de la asignación del sexo; se realiza en centros hospitalarios especializados10,17.
Las guías de práctica médica usualmente sugieren que las familias sean adecuadamente informadas de esta condición lo más pronto posible después del diagnóstico. Existen grupos internacionales de soporte bien establecidos para las personas con SIA10.
CONTRIBUCIÓN DE LOS AUTORES
Patricia Moreira Aguirre: Concepción y diseño del trabajo; recolección y obtención de resultados; análisis e interpretación de datos; redacción del manuscrito.
Conflicto de interés
Los autores declararon no tener ningún conflicto de interés personal, financiero, intelectual, económico y de interés corporativo con el Hospital Metropolitano y los miembros de la Revista MetroCiencia.
REFERENCIAS BIBLIOGRÁFICAS
Motos M, Ladrón de Guevara N, Salamanca A, Mozas J. Síndrome de insensibilidad androgénica. Grupo de trabajo sobre cáncer en síndromes genéticos polimalformativos. 2013. Disponible en: https://www.orpha.net/data/patho/Pro/es/Sindrome-Insensibilidad-Androgenica.pdf
Solana L, Paris A. Síndrome de insensibilidad completa a los andrógenos. Revista SAEGRE 2013;XX(1): 36-40. Disponible en: http://www.saegre.org.ar/revista/numeros/2013/n1/36-40-2013n1.pdf
Guerrero-Fernández J, Azcona C, Barreiro J, Bermúdez J, Carcavilla A, et al. Guía de actuación en las anomalías de la diferenciación sexual (ADS) / desarrollo sexual diferente (DSD). Anales Pediatría (Barcelona) 2018;89(5):315 e1-315 e19. https://doi.org/10.1016/j.anpedi.2018.06.009
Audi L, Bueno M, Calzada R, Cassorla F, Diéguez C, Ferrández, et al. Tratado de Endocrinología Pediátrica Pombo; 4ta edición. McGraw-Hill-Interamericana. Madrid-España 2009;122-132. 580-605. Disponible en: https://accessmedicina.mhmedical.com/book.aspx?bookID=1508
Beltrán O, Ramírez A, Garzón C, Orjuela C. Síndrome de insensibilidad androgénica debido a mutación intrónica en el gen del receptor androgénico. Presentación de un caso. Rev Med San 2016;19(2):116-120. Disponible en: https://www.unisanitas.edu.co/Revista/index.php?edicion=59&seccion=20
Hiort O. Pathogenesis and clinical features of disorders of androgen action 2019. Up to date. Disponible en: https://www.uptodate.com/contents/pathogenesis-and-clinical-features-of-disorders-of-androgen-action
Farias-Cortéz, J, Minakata-Ochoa F, Portillo-Sedano I. Síndrome de insensibilidad completa a los andrógenos: reporte de un caso, ilustración del manejo quirúrgico. Rev Mex Urol. 2014:74(2):117-122. Disponible en: https://biblat.unam.mx/en/revista/revista-mexicana-de-urologia/articulo/sindrome-de-insensibilidad-completa-a-los-androgenos-reporte-de-un-caso-ilustracion-del-manejo-quirurgico
Mclnerny T, Adam H, Campbell D, Kamat D, Kelleher K, Hoekelman R. Tratado de Pediatría. American Academy of Pediatric. Editorial Panamericana, Madrid, España. 2011; 2702-2710. Rodríguez-Estévez A, Grau G, Vela A, Rica I. Avances en el diagnóstico clínico, bioquímico y molecular de las 46,XY diferencias en el desarrollo sexual. Rev Esp Endocrinol Pediatr. 2015: 6(2). Disponible en: https://www.endocrinologiapediatrica.org/revistas/P1-E17/P1-E17-S736-A330.pdf
Hiort O. Diagnosis and treatment of disorders of the androgen receptor 2019. Up to date. Disponible en: https://www.uptodate.com/contents/diagnosis-and-treatment-of-disorders-of-the-androgen-receptor
Cha Y, Levitsky L. Causes of disorders of sex development. Up to date. 2019. Disponible en: https://www.uptodate.com/contents/causes-of-disorders-of-sex-development
Corbetta S, Muzza M, Avagliano L, Bulfamante G, Gaetty L, Eller-Vainicher C, et al. Gonadal structures in a fetus with complete androgen insensitivity syndrome and persistent Mullerian derivatives: comparison with normal fetal development. Fertil Steril 2011;95(3):1119e9-1119e14. https://doi.org/10.1016/j.fertnstert.2010.09.028
Bianca S, Cataliotti A, Bartoloni G, Torresnte I, Barrano B, Boemi G, et al. Prenatal diagnosis of androgen insentivity syndrome. Fetal Diagn Ther 2009;26(3):167-169. https://doi.org/10.1159/000251712
Sobel V, Schwartz B, Zhu Y, Cordero J, Imperato-MacGinley J. Bone mineral density in the complete androgen insensitivity and 5-alpha-reductase-2 deficiency syndromes. J Clin Endoc Metab 2006;91:3017-3023. https://doi.org/10.1210/jc.2005-2809
Dati E, Baroncelli G, Mora S, Russo G, Baldinotti F, Parrini D, Erba P, et al. Body composition and metabolic profile in women with complete androgen insensitivity syndrome. Sex Dev 2009;3(4):188-193. https://doi.org/10.1159/000228719
Wisniewski AB, Migeon C, Meyer-Bahlburg H, et al. Complete androgen insenstivity syndrome: long term medical, surgical, and psychosexual outcome. J Clin Endocrinol Metab 2000;85:2664-2669. https://doi.org/10.1210/jcem.85.8.6742
Barreiro J, Cabanas P, Angulo J, Castro-Feijoo L. Desarrollo sexual diferente (DSD): tratamiento farmacológico. Rev Esp Endocrinol Pediatr. 2015;6(2):21-27. Disponible en: https://www.endocrinologiapediatrica.org/revistas/P1-E17/P1-E17-S736-A322.pdf
Kohler B, Lumbroso S, Leger J, et al. Androgen insensitivity syndrome: somatic mosaicism of the androgen receptor in seven families and consequences for sex assignment and genetic counseling. J Clin Endocrinol Metab 2005;90:106-111. https://doi.org/10.1210/jc.2004-0462
Herman M, Wernikcke G, Yan W, Nori D, Parashar B. Pure seminoma in the setting of androgen insensitivity syndrome treated with surgical resectioin and para-aortic radiation: a case report and review of literatura. J Can Res Ther. 2010;6:318-320. https://doi.org/10.4103/0973-1482.73337
Dell´Edera D, Malvasi A, VItullo E, Epifania A, Tinelli A, Laterza E, et al. Androgen insenstivity syndrome (or Morris syndrome) and other associated pathologies. Eur Rev Med Pharmacol Sci 2010;14(11):947-957. Disponible en: https://www.europeanreview.org/article/843
Moreira P. Síndrome de insensibilidad androgénica. A propósito de un caso clínico. MetroCiencia. 2020 ene; 28(1): p. 20-24. DOI: https://doi.org/10.47464/MetroCiencia/vol28/1/2020/40-47. Available from: https://revistametrociencia.com.ec/index.php/revista/article/view/56